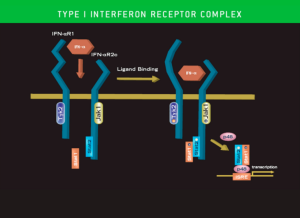
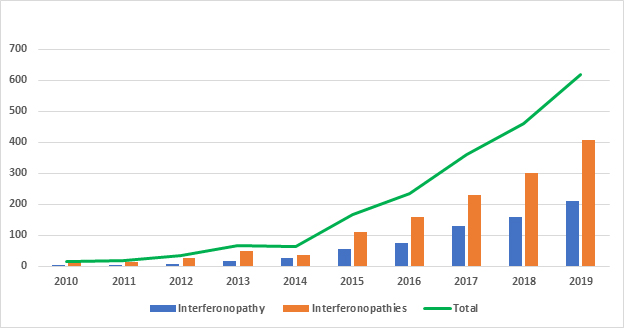
Figure 1: Recent Interferonopathy(ies) Publication Trend
Diseases Characterized by Interferon Overproduction
The connection between diseases characterized by IFN overproduction was initially made by Crow et al. when they identified phenotypic overlaps between Aicardi-Goutieres syndrome (AGS) and Cree encephalitis in 20031. AGS was originally labelled a Pseudo-TORCH syndrome prior to identification of its underlying genetic causes. Pseudo-TORCH syndrome is a group of disorders that mimic congenital TORCH (toxoplasmosis, rubella, cytomegalovirus, and herpes) infections, but test serologically negative. This autosomal recessive disorder is characterized by an increase in type I IFN and mutation of any of several nuclease genes, including TREX1, RNASEH2A, RNASEH2B, and RNASEH2C. It has been shown that these mutations result in an accumulation of DNA and RNA fragments generated during transcription, replication, DNA repair, and cell death. As a result, the nucleotide-sensing machinery of the immune system is activated resulting in marked production of type I IFNs and induction of the downstream pathologies now known as interferonopathy3. The importance of this finding is underscored by the other diseases that share overproduction of type I IFN as a characteristic. These include autoimmune diseases of unknown etiology, such as systemic lupus erythematosus (SLE), in which mutations in genes associated with nucleic acid metabolism have also been identified. Diseases such as type I diabetes, rheumatoid arthritis, Sjögrens syndrome, dermatomyositis (DM), and multiple sclerosis (MS) also exhibit strong type I IFN signatures.
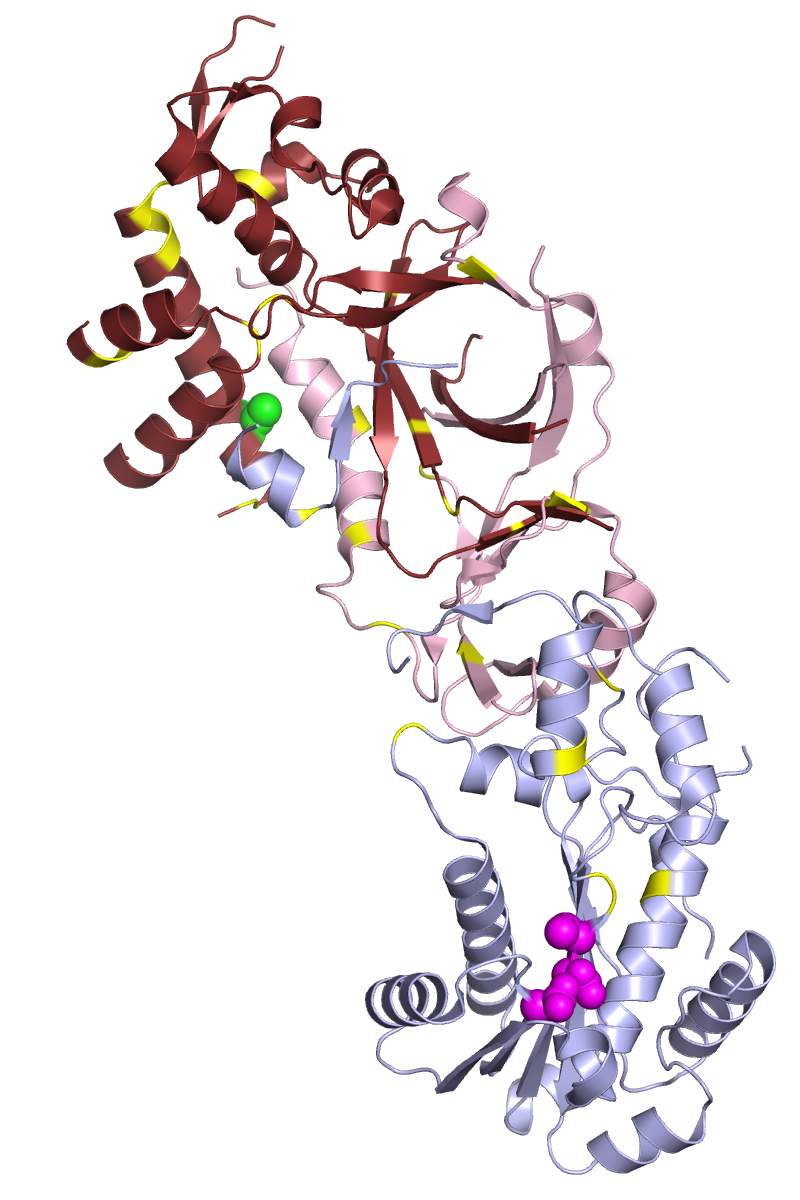
Figure 2:The Human Ribonuclease H2 Complex. The positions of residues whose mutation is associated with AGS are indicated in yellow
(adapted under GNU Free documentation license https://commons.wikimedia.org/wiki/File:3puf_rnaseH2_AGS_mutations.png)
Mutations in Nucleic Acid Sensing Machinery
While the nucleic acid sensing ability of the innate immune system is central to the pathogen response of the host, it also poses a threat due to the fact that host DNA and RNA can cause the unwanted activation of this system. Because all cytosolic nucleic acid sensing pathways induce IFN production, it is not surprising that several interferonopathies have been linked to mutations in this machinery. For example, gain-of-function mutations in MDA5 (IFIH1), RIG-I (DDX58), and STING (TMEM173) all result in increased type I IFN production. Both retinoic acid-inducible gene 1 (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) sense small RNA molecules, typically produced by viruses, to induce activation of the antiviral type I and type III interferons, while STING senses intracellular DNA, which can be of bacterial origin4. During normal innate immune responses, these nucleotide sensors help rid cells of invading pathogens. However, in interferonopathies the dysregulated production of pleiotropic type I IFNs can result in tissue damage5.
The remarkable number of the pathways leading to production of type I IFN affords the innate immune system to ability to respond to a multitude of pathogens. However, it also complicates the identification of the underlying cause of diseases in which IFN is overproduced. Thus, in many cases the ability to identify the cellular source of IFN with precise and sensitive assays is necessary to determine the mechanism underlying IFN overproduction.
Known Interferonopathies
Diseases which have been characterized as type I interferonopathies include AGS, spondyloenchondrodysplasia with immune dysregulation, stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy (SAVI), Japanese autoinflammatory syndrome with lipodystrophy (JASL), ubiquitin-specific peptidase 18 deficiency, chronic atypical neutrophilic dermatitis with lipodystrophy, DNA II deficiency, and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE)3. Most of these are rare diseases and not all of their phenotypic symptoms overlap. Further, as new mutations are identified and the inclusion criteria for interferonopathies becomes more definitive this list has changed. Nonetheless, the important work done to uncover the cause of the symptom’s characteristic of interferonopathies has propelled the field toward the identification of therapeutics and even initial clinical trials for some of these debilitating diseases. Loss of function mutants identified as interferonopathies include mutations in USP18 and ISG15. USP18 is a deubiquitinating protease important in the downregulation of key molecules involved in type I IFN signaling. One important function of USP18 is the removal of ISG15 additions from proteins. ISG15 is a ubiquitin-like protein with broad involvement in the host antiviral response6,7. Data suggests that the ISG15 protein broadly targets newly synthesized proteins for degradation. This coincides with its known antiviral activities as it is believed to target newly-synthesized viral proteins8. Further, intracellular ISG15 is believed to prevent overamplification of the type I IFN response and in its absence USP18 levels are also decreased, although the precise mechanisms underlying this remain to be determined9.
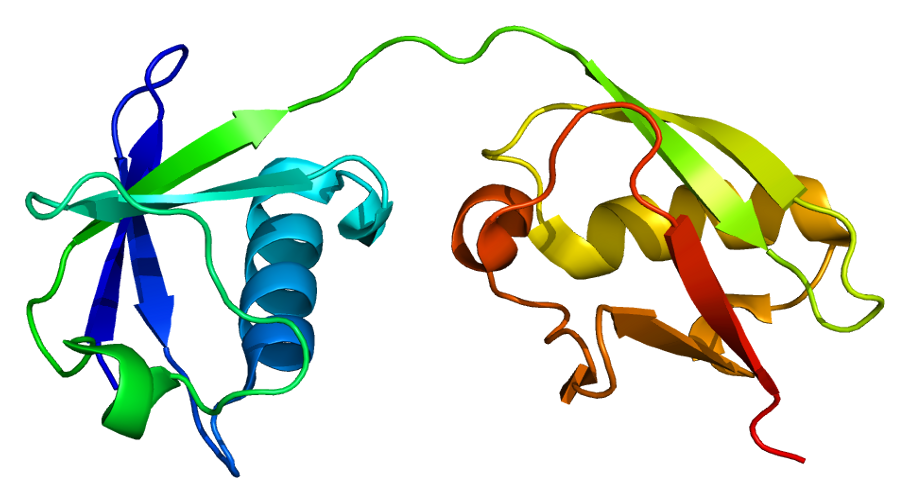
Figure 3:ISG15 Protein Structure
(https://commons.wikimedia.org/wiki/File:Protein_ISG15_PDB_1z2m.png)
Autoimmune Diseases and Overproduction of Type I IFN
While many of the interferonopathies described above can be considered congenital in nature, with symptoms appearing at or shortly after birth, autoimmune diseases such as SLE appear later in life; however, there is often a genetic component of predisposition to these diseases. Moreover, the production of autoantibodies against nuclei acids is a hallmark of SLE and can appear years before other symptoms occur. The origin of nucleic acids in this disease remains unclear and may vary between patients, but likely candidates include increased damage or death of cells and defective nucleic acid metabolism. Nonetheless, the result is an accumulation of nucleic acids that may activate the same pathways involved in interferonopathies and be the cause for the overproduction of IFN10. The difficulty identifying the main causes of diseases such as SLE may be due to the range of underlying mutations. This necessitates the ability to identify cellular sources of IFN, which could also be useful in tailoring of individualized medicine for patients. Sensitive assays to monitor IFN overproduction could also be useful when identifying likely responders and non-responders to individual therapeutic approaches.
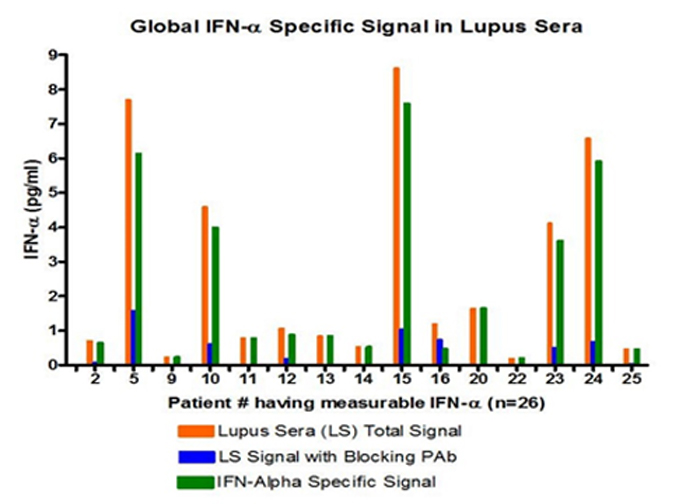
Figure 4:Global IFN-Alpha Specific signal in Lupus Sera
Therapeutics for Autoimmune Diseases and Interferonopathies
Because all interferonopathies result in overproduction of type I IFN, IFN itself, the type I IFNAR, and downstream signaling molecules are all potential therapeutic targets. For example, anti-interferon therapy is being explored for treatment of SLE11. First generation Janus kinase inhibitors, including baricitinib, tofacitinib and ruxolitinib, are currently in clinical trials and some have shown positive results in treatment of asthma and lupus12. Moreover, current clinical trials are testing reverse transcriptase inhibitors for treatment of Aicardi-Goutières syndrome [ClinicalTrials.gov Identifier: NCT03304717, Clinical- Trials.gov Identifier: NCT02363452 ].
Methods for Studying Interferonopathies and Identifying Novel Therapeutics
Confirmation of the hypothesis that overproduction of type I IFNs (IFN Alpha, IFN Beta, and IFN Omega) is causative of interferonopathies, and not simply a correlation, has been shown through initial trials of IFN pathway-specific therapeutics. In general, the inclusion criteria for interferonopathies has been somewhat broad and typically includes measurement of the upregulation of interferon-stimulated genes (ISGs) or other biomarkers indicative of activation of this pathway. Highly sensitive (low pg/ml) or ultra-sensitive (fg/ml) assays for IFN Alpha, Beta and Omega quantitation are therefore needed to further establish the link between type I IFNs overproduction and interferonopathies. In addition to sensitivity, an IFN alpha assay would ideally be able to measure All IFN alpha subtypes in order to provide a more complete picture on the role of different IFN alpha subtypes in interferonopathies.
Ultra-sensitive biomarker assays such as Single Molecule Array (SIMOA) assays, would also help pave the way to further understand the underlining mechanism of interferonopathies. Furthermore, assays that can reliably reflect brain injuries such as those seen in interferonopathies have long been sought. Recent publications have highlighted the correlation between neurofilament light chain (NFL) in the blood, serum, or CNS and brain injury caused by traumatic injury, untreated HIV-1 infection, or multiple sclerosis (MS)13. Thus, the NF-L assay on SIMOA platform may prove beneficial in the study of interferonopathies, especially for purposes of early disease monitoring.
References
1. Crow YJ, Black DN, Ali M, et al. Cree encephalitis is allelic with aicardi-goutières syndrome: Implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet. 2003. doi:10.1136/jmg.40.3.183
2. Ali M, Highet LJ, Lacombe D, et al. A second locus for Aicardi-Goutières syndrome at chromosome 13q14-21. J Med Genet. 2006. doi:10.1136/jmg.2005.031880
3. Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. 2015. doi:10.1038/nri3850
4. Wu J, Chen ZJ. Innate Immune Sensing and Signaling of Cytosolic Nucleic Acids. Annu Rev Immunol. 2014. doi:10.1146/annurev-immunol-032713-120156
5. Uggenti C, Lepelley A, Crow YJ. Self-Awareness: Nucleic Acid–Driven Inflammation and the Type I Interferonopathies. Annu Rev Immunol. 2019. doi:10.1146/annurev-immunol-042718-041257
6. Honke N, Shaabani N, Zhang DE, Hardt C, Lang KS. Multiple functions of USP18. Cell Death Dis. 2016. doi:10.1038/cddis.2016.326
7. Perng YC, Lenschow DJ. ISG15 in antiviral immunity and beyond. Nat Rev Microbiol. 2018. doi:10.1038/s41579-018-0020-5
8. Durfee LA, Lyon N, Seo K, Huibregtse JM. The ISG15 Conjugation System Broadly Targets Newly Synthesized Proteins: Implications for the Antiviral Function of ISG15. Mol Cell. 2010. doi:10.1016/j.molcel.2010.05.002
9. Zhang X, Bogunovic D, Payelle-Brogard B, et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature. 2015. doi:10.1038/nature13801
10. Ghodke-Puranik Y, Niewold TB. Immunogenetics of systemic lupus erythematosus: A comprehensive review. J Autoimmun. 2015. doi:10.1016/j.jaut.2015.08.004
11. Kirou KA, Gkrouzman E. Anti-interferon alpha treatment in SLE. Clin Immunol. 2013. doi:10.1016/j.clim.2013.02.013
12. Crow MK, Rönnblom L. Report of the inaugural Interferon Research Summit: Interferon in inflammatory diseases. In: Lupus Science and Medicine. ; 2018. doi:10.1136/lupus-2018-000276
13. Gisslén M, Price RW, Andreasson U, et al. Plasma Concentration of the Neurofilament Light Protein (NFL) is a Biomarker of CNS Injury in HIV Infection: A Cross-Sectional Study. EBioMedicine. 2016. doi:10.1016/j.ebiom.2015.11.036
Diseases Characterized by Interferon Overproduction
The connection between diseases characterized by IFN overproduction was initially made by Crow et al. when they identified phenotypic overlaps between Aicardi-Goutieres syndrome (AGS) and Cree encephalitis in 20031. AGS was originally labelled a Pseudo-TORCH syndrome prior to identification of its underlying genetic causes. Pseudo-TORCH syndrome is a group of disorders that mimic congenital TORCH (toxoplasmosis, rubella, cytomegalovirus, and herpes) infections, but test serologically negative. This autosomal recessive disorder is characterized by an increase in type I IFN and mutation of any of several nuclease genes, including TREX1, RNASEH2A, RNASEH2B, and RNASEH2C. It has been shown that these mutations result in an accumulation of DNA and RNA fragments generated during transcription, replication, DNA repair, and cell death. As a result, the nucleotide-sensing machinery of the immune system is activated resulting in marked production of type I IFNs and induction of the downstream pathologies now known as interferonopathy3. The importance of this finding is underscored by the other diseases that share overproduction of type I IFN as a characteristic. These include autoimmune diseases of unknown etiology, such as systemic lupus erythematosus (SLE), in which mutations in genes associated with nucleic acid metabolism have also been identified. Diseases such as type I diabetes, rheumatoid arthritis, Sjögrens syndrome, dermatomyositis (DM), and multiple sclerosis (MS) also exhibit strong type I IFN signatures.
Mutations in Nucleic Acid Sensing Machinery
While the nucleic acid sensing ability of the innate immune system is central to the pathogen response of the host, it also poses a threat due to the fact that host DNA and RNA can cause the unwanted activation of this system. Because all cytosolic nucleic acid sensing pathways induce IFN production, it is not surprising that several interferonopathies have been linked to mutations in this machinery. For example, gain-of-function mutations in MDA5 (IFIH1), RIG-I (DDX58), and STING (TMEM173) all result in increased type I IFN production. Both retinoic acid-inducible gene 1 (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) sense small RNA molecules, typically produced by viruses, to induce activation of the antiviral type I and type III interferons, while STING senses intracellular DNA, which can be of bacterial origin4. During normal innate immune responses, these nucleotide sensors help rid cells of invading pathogens. However, in interferonopathies the dysregulated production of pleiotropic type I IFNs can result in tissue damage5.
The remarkable number of the pathways leading to production of type I IFN affords the innate immune system to ability to respond to a multitude of pathogens. However, it also complicates the identification of the underlying cause of diseases in which IFN is overproduced. Thus, in many cases the ability to identify the cellular source of IFN with precise and sensitive assays is necessary to determine the mechanism underlying IFN overproduction.
Known Interferonopathies
Diseases which have been characterized as type I interferonopathies include AGS, spondyloenchondrodysplasia with immune dysregulation, stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy (SAVI), Japanese autoinflammatory syndrome with lipodystrophy (JASL), ubiquitin-specific peptidase 18 deficiency, chronic atypical neutrophilic dermatitis with lipodystrophy, DNA II deficiency, and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE)3. Most of these are rare diseases and not all of their phenotypic symptoms overlap. Further, as new mutations are identified and the inclusion criteria for interferonopathies becomes more definitive this list has changed. Nonetheless, the important work done to uncover the cause of the symptom’s characteristic of interferonopathies has propelled the field toward the identification of therapeutics and even initial clinical trials for some of these debilitating diseases. Loss of function mutants identified as interferonopathies include mutations in USP18 and ISG15. USP18 is a deubiquitinating protease important in the downregulation of key molecules involved in type I IFN signaling. One important function of USP18 is the removal of ISG15 additions from proteins. ISG15 is a ubiquitin-like protein with broad involvement in the host antiviral response6,7. Data suggests that the ISG15 protein broadly targets newly synthesized proteins for degradation. This coincides with its known antiviral activities as it is believed to target newly-synthesized viral proteins8. Further, intracellular ISG15 is believed to prevent overamplification of the type I IFN response and in its absence USP18 levels are also decreased, although the precise mechanisms underlying this remain to be determined9.
(https://commons.wikimedia.org/wiki/File:Protein_ISG15_PDB_1z2m.png)
Autoimmune Diseases and Overproduction of Type I IFN
While many of the interferonopathies described above can be considered congenital in nature, with symptoms appearing at or shortly after birth, autoimmune diseases such as SLE appear later in life; however, there is often a genetic component of predisposition to these diseases. Moreover, the production of autoantibodies against nuclei acids is a hallmark of SLE and can appear years before other symptoms occur. The origin of nucleic acids in this disease remains unclear and may vary between patients, but likely candidates include increased damage or death of cells and defective nucleic acid metabolism. Nonetheless, the result is an accumulation of nucleic acids that may activate the same pathways involved in interferonopathies and be the cause for the overproduction of IFN10. The difficulty identifying the main causes of diseases such as SLE may be due to the range of underlying mutations. This necessitates the ability to identify cellular sources of IFN, which could also be useful in tailoring of individualized medicine for patients. Sensitive assays to monitor IFN overproduction could also be useful when identifying likely responders and non-responders to individual therapeutic approaches.
Therapeutics for Autoimmune Diseases and Interferonopathies
Because all interferonopathies result in overproduction of type I IFN, IFN itself, the type I IFNAR, and downstream signaling molecules are all potential therapeutic targets. For example, anti-interferon therapy is being explored for treatment of SLE11. First generation Janus kinase inhibitors, including baricitinib, tofacitinib and ruxolitinib, are currently in clinical trials and some have shown positive results in treatment of asthma and lupus12. Moreover, current clinical trials are testing reverse transcriptase inhibitors for treatment of Aicardi-Goutières syndrome [ClinicalTrials.gov Identifier: NCT03304717, Clinical- Trials.gov Identifier: NCT02363452 ].
Methods for Studying Interferonopathies and Identifying Novel Therapeutics
Confirmation of the hypothesis that overproduction of type I IFNs (IFN Alpha, IFN Beta, and IFN Omega) is causative of interferonopathies, and not simply a correlation, has been shown through initial trials of IFN pathway-specific therapeutics. In general, the inclusion criteria for interferonopathies has been somewhat broad and typically includes measurement of the upregulation of interferon-stimulated genes (ISGs) or other biomarkers indicative of activation of this pathway. Highly sensitive (low pg/ml) or ultra-sensitive (fg/ml) assays for IFN Alpha, Beta and Omega quantitation are therefore needed to further establish the link between type I IFNs overproduction and interferonopathies. In addition to sensitivity, an IFN alpha assay would ideally be able to measure All IFN alpha subtypes in order to provide a more complete picture on the role of different IFN alpha subtypes in interferonopathies.
Ultra-sensitive biomarker assays such as Single Molecule Array (SIMOA) assays, would also help pave the way to further understand the underlining mechanism of interferonopathies. Furthermore, assays that can reliably reflect brain injuries such as those seen in interferonopathies have long been sought. Recent publications have highlighted the correlation between neurofilament light chain (NFL) in the blood, serum, or CNS and brain injury caused by traumatic injury, untreated HIV-1 infection, or multiple sclerosis (MS)13. Thus, the NF-L assay on SIMOA platform may prove beneficial in the study of interferonopathies, especially for purposes of early disease monitoring.
References
1. Crow YJ, Black DN, Ali M, et al. Cree encephalitis is allelic with aicardi-goutières syndrome: Implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet. 2003. doi:10.1136/jmg.40.3.183
2. Ali M, Highet LJ, Lacombe D, et al. A second locus for Aicardi-Goutières syndrome at chromosome 13q14-21. J Med Genet. 2006. doi:10.1136/jmg.2005.031880
3. Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. 2015. doi:10.1038/nri3850
4. Wu J, Chen ZJ. Innate Immune Sensing and Signaling of Cytosolic Nucleic Acids. Annu Rev Immunol. 2014. doi:10.1146/annurev-immunol-032713-120156
5. Uggenti C, Lepelley A, Crow YJ. Self-Awareness: Nucleic Acid–Driven Inflammation and the Type I Interferonopathies. Annu Rev Immunol. 2019. doi:10.1146/annurev-immunol-042718-041257
6. Honke N, Shaabani N, Zhang DE, Hardt C, Lang KS. Multiple functions of USP18. Cell Death Dis. 2016. doi:10.1038/cddis.2016.326
7. Perng YC, Lenschow DJ. ISG15 in antiviral immunity and beyond. Nat Rev Microbiol. 2018. doi:10.1038/s41579-018-0020-5
8. Durfee LA, Lyon N, Seo K, Huibregtse JM. The ISG15 Conjugation System Broadly Targets Newly Synthesized Proteins: Implications for the Antiviral Function of ISG15. Mol Cell. 2010. doi:10.1016/j.molcel.2010.05.002
9. Zhang X, Bogunovic D, Payelle-Brogard B, et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature. 2015. doi:10.1038/nature13801
10. Ghodke-Puranik Y, Niewold TB. Immunogenetics of systemic lupus erythematosus: A comprehensive review. J Autoimmun. 2015. doi:10.1016/j.jaut.2015.08.004
11. Kirou KA, Gkrouzman E. Anti-interferon alpha treatment in SLE. Clin Immunol. 2013. doi:10.1016/j.clim.2013.02.013
12. Crow MK, Rönnblom L. Report of the inaugural Interferon Research Summit: Interferon in inflammatory diseases. In: Lupus Science and Medicine. ; 2018. doi:10.1136/lupus-2018-000276
13. Gisslén M, Price RW, Andreasson U, et al. Plasma Concentration of the Neurofilament Light Protein (NFL) is a Biomarker of CNS Injury in HIV Infection: A Cross-Sectional Study. EBioMedicine. 2016. doi:10.1016/j.ebiom.2015.11.036
Related Article
Performance Characterization of a High-Sensitivity Human IFN-Beta ELISA in Healthy Serum, Patient Serum, and Plasma Samples
Read More