Summary: This article examines how Type I IFNs can support or undermine peripheral immune tolerance by shaping regulatory T cells, epigenetic programs, and thymus selection, and why dysregulated signaling is linked to autoimmunity.
Introduction
The immune system walks a perpetual tightrope. On one side lies the danger of infection and malignancy; on the other, the peril of self-destruction through autoimmunity. Peripheral immune tolerance — the set of mechanisms that silence autoreactive lymphocytes escaping the thymus — is one of the most critical balancing acts in biology. Within this landscape, type I interferons (IFNs) occupy a paradoxical position: they are simultaneously architects of self-tolerance and, when dysregulated, engines of autoimmune disease. Understanding their role has become one of the most compelling frontiers in modern immunology.
What are Type I Interferons
Type I IFNs are a family of cytokines — principally IFN-α and IFN-β — that signal through a shared heterodimeric receptor, IFNAR (composed of IFNAR1 and IFNAR2). Classically induced by viral infection and other pathogen-associated molecular patterns, they activate Janus kinase–signal transducer and activator of transcription (JAK-STAT) signaling, driving transcription of hundreds of interferon-stimulated genes (ISGs). Yet it is increasingly clear that type I IFNs are not simply antiviral alarm signals — they are pleiotropic cytokines that touch nearly every corner of the adaptive immune system. As a comprehensive 2024–2025 review in Physiological Reviews notes, type I IFNs play multifaceted antiviral, immunomodulatory, and antitumor roles while also contributing to a spectrum of autoimmune disorders [1].
The Tolerogenic Foundation: What Peripheral Tolerance Actually Requires
Central tolerance in the thymus eliminates most overtly self-reactive T cells, but it is imperfect. A residual pool of potentially autoreactive lymphocytes circulates in every individual. Peripheral tolerance mechanisms — including T cell anergy, clonal deletion, regulatory T cell (Treg) suppression, and immune checkpoint engagement — continuously silence these cells outside the thymus. Dendritic cells (DCs) are central orchestrators of these processes, presenting self-antigens in the absence of co-stimulation to induce anergy or deletion, or actively converting naïve T cells into Foxp3+ Tregs [2].
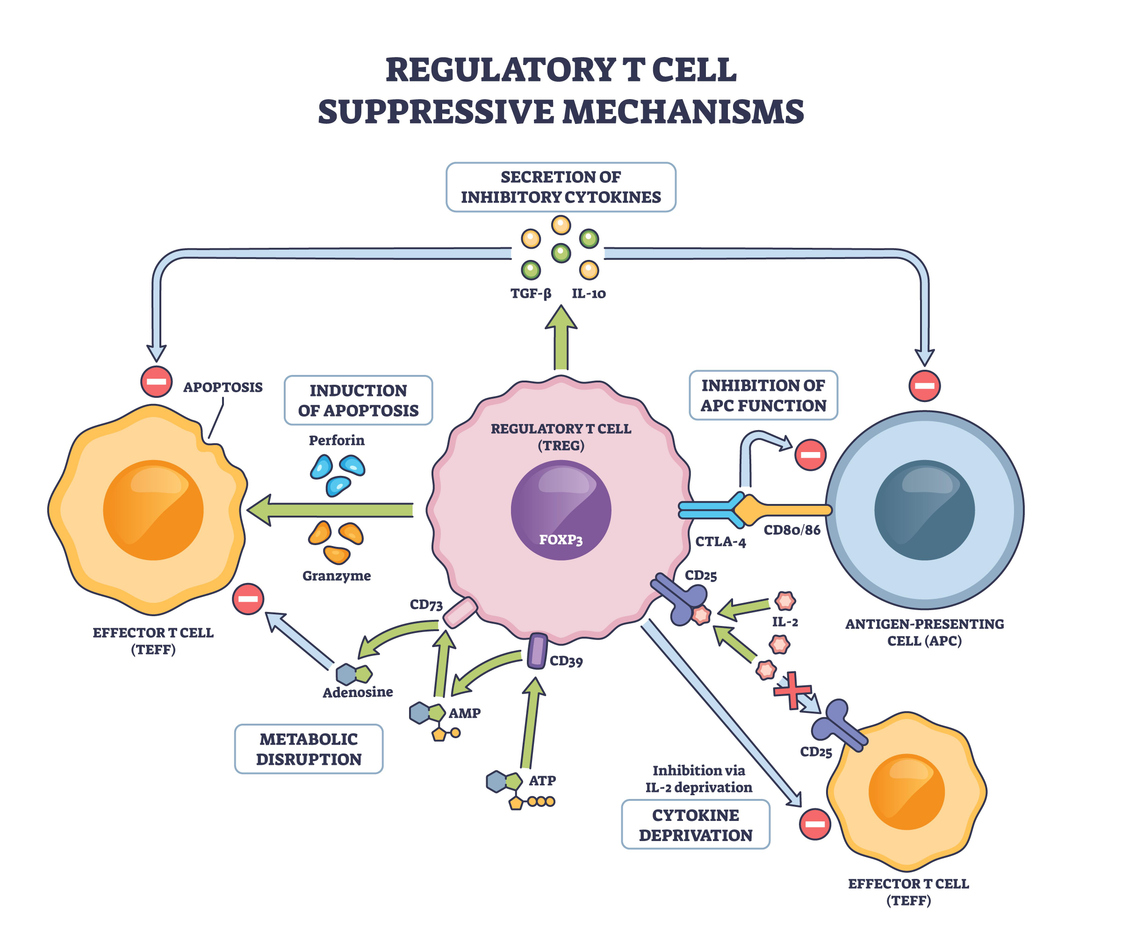
Type I IFNs and regulatory T Cell Biology: A Doube-Edged Sword
Tregs are perhaps the most pivotal effectors of peripheral tolerance. They express Foxp3 and suppress autoreactive lymphocytes through contact-dependent inhibition and anti-inflammatory cytokines such as IL-10 and TGF-β. Type I IFNs act on Tregs in ways that are context-dependent and sometimes contradictory.
On the tolerogenic side, a landmark 2022 Immunity study demonstrated that IFN-β acts directly on naive CD4+ T cells to promote Foxp3 acetylation via STAT1-driven upregulation of the acetyltransferase P300, thereby enhancing Treg induction. Critically, this mechanism was validated in human PBMCs — providing direct translational relevance — and was shown to prolong allograft survival [3]. These findings mechanistically explain a long-observed clinical phenomenon: IFN-β treatment in relapsing-remitting multiple sclerosis consistently increases peripheral blood Treg frequency.
IFNAR signaling also appears important for optimal Treg survival and fitness. Studies using mixed bone marrow chimeras showed that IFNAR-deficient Tregs express elevated levels of the pro-apoptotic molecule Bim and display reduced phospho-STAT5, a key survival signal downstream of IL-2 [4]. Without type I IFN input, Tregs adopt a more naïve, less activated phenotype and are underrepresented in secondary lymphoid organs — a deficit that can be partially rescued by IL-2 immunocomplexes.
Work in the experimental autoimmune encephalomyelitis (EAE) model added further nuance. Conditional deletion of IFNAR specifically in Foxp3+ Tregs results in significantly more severe EAE with earlier onset, despite those Tregs displaying a paradoxically more activated phenotype. The mechanistic explanation is that type I IFN signaling in Tregs normally drives chemokine production (CCL8, CCL9, CCL22) that recruits myeloid-derived suppressor cells (MDSCs) to draining lymph nodes early in the priming phase — a multi-layered suppression network that collapses when IFNAR is absent from Tregs [5].
Chromatin, Epigenetic, and Innate Memory Tolerance
Beyond direct Treg effects, type I IFNs regulate peripheral tolerance at the epigenetic level. A 2024 Immunological Reviews paper by Mishra and Ivashkiv elegantly described how interferons play key roles in training, priming, and tolerance of monocytes and hematopoietic progenitors through chromatin-mediated mechanisms. Rather than simply activating ISGs, IFNs remodel chromatin at regulatory elements of inflammatory genes — including TNF, IL6, and IL1B — to establish tolerant states that dampen secondary responses [6]. This form of epigenetic silencing, when properly calibrated, prevents hyperinflammation and protects peripheral tissues from collateral damage.
Dysregulation of this same system is a hallmark of autoimmunity. In systemic lupus erythematosus (SLE), chronic high-level type I IFN signaling is associated with DNA hypomethylation at ISG loci in CD4+ T cells, creating a self-perpetuating epigenetic sensitization that amplifies IFN responses and breaks tolerance [7]. The 2025 MDPI review in Biomolecules on the type I IFN axis in systemic autoimmune diseases confirms that a substantial proportion of SLE-risk variants are associated with IFN-I production or signaling — cementing type I IFNs as central players in tolerance breakdown [8].
The Thymus-Periphery Axis: Setting the Stage for Peripheral Tolerance
A remarkable 2024 Science Immunology study by Ashby, Vobořil, and colleagues revealed that type I and type III IFNs are constitutively produced by a tiny subset of AIRE-expressing medullary thymic epithelial cells (mTECs) — independently of infection. These thymic IFNs promote DC1 and macrophage maturation, and critically, their loss reduced regulatory T cell selection and diminished T cell receptor (TCR) repertoire diversity, leading to enhanced autoreactive T cell responses against ISG-derived self-antigens expressed during peripheral IFN signaling [9]. This finding draws a direct mechanistic thread between thymic IFN production and the quality of peripheral tolerance: if T cells are not tolerized to ISG-encoded self-antigens in the thymus, they can become autoreactive when peripheral IFN is induced.
When Tolerance Fails: Type I IFNs in Autoimmune Disease
The dark side of type I IFN biology in peripheral tolerance is most vivid in SLE and related interferonopathies. In SLE, chronic activation of plasmacytoid DCs (pDCs) drives excess IFN-α production, which promotes DC maturation, autoreactive B cell differentiation into plasmablasts, Th17 skewing, and Treg dysfunction [10]. A 2024 Frontiers in Immunology article on type I IFNs in lupus nephritis summarized how IFN-I both initiates and perpetuates tolerogenic breakdown — promoting autoantibody generation, immune complex deposition, and complement activation in a self-reinforcing loop [11].
Interestingly, not all IFN-I activity in peripheral tissues is pathogenic. The same review notes that gut IFN-α can be protective by inducing IL-27 in DCs and sustaining Foxp3+ Tregs to maintain mucosal immune tolerance [11] — yet another illustration of the exquisite context-dependency of type I IFN biology.
Therapeutic Implications
The therapeutic manipulation of type I IFN pathways to restore peripheral tolerance is an active area of clinical development. Anifrolu-mab, a monoclonal antibody targeting IFNAR1, has received FDA approval for SLE and demonstrated efficacy by broadly blocking type I IFN signaling. JAK inhibitors, which suppress downstream STAT signaling, represent another avenue. Meanwhile, the IFN-β/Foxp3 acetylation axis identified in the Immunity 2022 study suggests that low-dose type I IFN strategies — rather than pan-blockade — might be harnessed to specifically boost Treg induction in transplantation or autoimmunity settings [3, 8].
Conclusion
Type I interferons are not merely antiviral cytokines — they are fundamental architects of the peripheral tolerance landscape. Through effects on Treg survival, induction, and function; through epigenetic chromatin remodeling; and through modulating DC behavior in both the thymus and periphery, they help maintain the fragile peace between immunity and self-tolerance. Yet their chronic dysregulation — in SLE, interferonopathies, and other systemic autoimmune diseases — can shatter that peace. The challenge for the next decade of immunology is to learn how to selectively exploit their tolerogenic potential without triggering their autoimmune consequences.
References
- Mavagani C.P. and Crow M.K.. Type I interferons in health and disease: molecular aspects and clinical implications. Physiological Reviews. 2024. https://journals.physiology.org/doi/abs/10.1152/physrev.00047.2024
- Parker V.K. et al. Tolerogenic Dendritic Cells for Regulatory T Cell Induction in Man. Frontiers in Immunology. 2015;6:569. https://pmc.ncbi.nlm.nih.gov/articles/PMC4638142/
- Fueyo-Gonzalez F., et al. Interferon-β acts directly on T cells to prolong allograft survival by enhancing regulatory T cell induction through Foxp3 acetylation. Immunity. 2022;55(2):247–261. https://www.cell.com/immunity/fulltext/S1074-7613(22)00040-1
- Metidji A, et al. IFN-α/β Receptor Signaling Promotes Regulatory T Cell Development and Function under Stress Conditions. The Journal of Immunology. 2015;194(9):4265–4276. https://journals.aai.org/jimmunol/article/194/9/4265/109115/
- Tanwar S, et al. Type I IFN signaling in T regulatory cells modulates chemokine production and Myeloid Derived Suppressor Cells trafficking during EAE. Journal of Autoimmunity. 2020;115:102520. https://pmc.ncbi.nlm.nih.gov/articles/PMC7712497/
- Mishra B, Ivashkiv LB. Interferons and epigenetic mechanisms in training, priming and tolerance of monocytes and hematopoietic progenitors. Immunological Reviews. 2024;323(1):257–275. https://pubmed.ncbi.nlm.nih.gov/38567833/
- Fakour P, et al. Epigenetic regulation of FOXP3 gene expression in relation to impaired function of regulatory T cells in systemic lupus erythematosus. Exploration of Immunology. 2024;4:640–657. https://www.explorationpub.com/Journals/ei/Article/1003164
- Ishihara R, et al. The Type I Interferon Axis in Systemic Autoimmune Diseases: From Molecular Pathways to Targeted Therapy. Biomolecules. 2025;15(11):1586. https://www.mdpi.com/2218-273X/15/11/1586
- Ashby KM, et al. Sterile Production of Interferons in the Thymus Impacts T Cell Repertoire Selection. Science Immunology. 2024;9(97):eadp1139. https://pmc.ncbi.nlm.nih.gov/articles/PMC12052003/
- Wang X, et al. Recent Advances of Type I Interferon on the Regulation of Immune Cells and the Treatment of Systemic Lupus Erythematosus. Journal of Inflammation Research. 2025;18:4533–4549. https://pubmed.ncbi.nlm.nih.gov/40182060/
- Lai B, et al. Therapeutically targeting proinflammatory type I interferons in systemic lupus erythematosus: efficacy and insufficiency with a specific focus on lupus nephritis. Frontiers in Immunology. 2024;15:1489205. https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1489205/full